
My little Sketch
Current Projects
There will be no new projects involving the use of dangerous chemicals. This page is now a relic.
Sodium Production
Electrochemical Lithium Production
Hydrazine Production
Ferrates
Preparation of THF using OTC Pentoses as the starting material.
- Chemical Reduction
Rxn #1
6NaOH + 4Al ---> 6Na + 3H2 + 2Al2O3
Rxn #2
4NaOH + 2Al + 2C ---> 4Na + 2CO + Al2O3
The first reaction is complicated by the competing reaction that wants to produce the aluminate and therefore reduces yields. My attempt to compensate for that is with the second reaction, knowing that aluminates are reduced with carbon at high temp my reasoning is that by incorporating the carbon into the mix it will reduce any aluminate formed via the heat of the initial thermite like reaction. Reaction 2 is actually two other combined reactions, the aluminate being decomposed and therefore being an intermediate product. Ideally I would like to run my reaction at 10 different points corresponding to each of the ten points on the above graph and superimpose my yields over the %rxn. However knowing my time constraints I will in all likelihood just perform a 100% Rxn 2, a 100% rxn. 1 and a 50-50 and connect the dots to get a gestimation of what combination is the best. You can see a picture of my sodium producing apparatus here.
-Electrochemical from NaCl
I'm starting to really like this idea. A melt of 50% CaCl2 and 50% NaCl has a melting point of near 550C which is close to what I work with in terms of a hydroxide melt. Additionally the Downs cell is less fickle, current density, electrode distance, and temperature play smaller roles. My plan is to insert the electrodes in a pot coming up from the bottom, the cell divided in the middle by a iron plate down to just above the electrodes so the cell doesn't short out. The chlorine side vented out and through copper tubing and through strongly basified water. The sodium side without gas outlet. The cover affixed on so it can't be removed, electrolysis would be begun and continued for a set amount of time then the heating and electrolysis stopped and the exit of gasses sealed which will create a vacuum. Once cool the vacuum is cracked by allowing mineral oil into the cell which should cover the sodium formed then it is taken out of the cell and everything examined. At least that is the plan.
My little Sketch
As can be read off the diagram the normal cell composition is 40% NaCl and 60% CaCl2 with a melting point of 575 - 585 C, as the reaction continues it consumes the NaCl and as the NaCl % drops the melting point drops as it gets closet to the system eutectic of 33% NaCl which has a melting point of 505C and from there the temperature should rise as the eutectic point becomes more distant. So I figure the whole system can be closed and not checked on, as long as I keep the temperature at about 600C things should work out for quite some time. To do this I need to purchase a K type thermocouple and an instrument to read the temperature it measures, I will insert this into a pipe that will extend in from the top and be sealed at the bottom containing copper BB's so it conducts the heat of the melt to the thermocouple for a good reading of the temperature without damaging the thermocouple. I also recently purchased a set of graphite electrodes for this cell, good ones, not battery ones, so they should last. The chlorine gas lead away will go somewhere, maybe into a hydroxide solution, I always wish I had a way to store it though. Got two good quality graphite rods, that's a start right?
Edit: 3/20/05
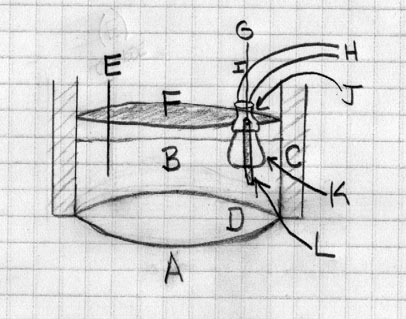
(A) Heating applied from beneath (B) Electrolyte, most likely the same as described above. (C) Insulation, fiberglass or something else like a refractory material. (D) The vessel itself, probably just a cast iron pan. (E) Cathode, graphite (F) Lid, probably just the one that comes with the pan, possibly with the bottom covered in a silicate based putty like fire place mortar to make it more inert. (G) Wire that goes in through a hole in the copper pipe (H) and bound in place at point (I) With silicate cement with care taken to ensure no connection with the copper pipe is made (J) Is a copper enlarging adapter on the inside of the lid and pulled into place to give a good seal and held there with more of the mortar. This is bound into the inverted porcelain crucible (K) That acts to funnel the Cl2 gas produced through pipe (H) Another graphite electrode (L) this one functioning as the anode is attached to the wire (G) as described previously. In this way the anode in insulated in the upper section by the crucible and chlorine gas produced should be neatly funneled up. The current running through the cell is determined initially after the contents are molten and the cell is run for one hour and the yield determined by allowing to cool then opening.
Normally I shy away from energetic materials, not only are they hazardous for your health but they have a tendency to attract law enforcement attention and are for the most part illegal. However this compound presents the opportunity for real research. Although I have found the triazide of borazine I have found neither hide nor hair of the trinitro compound. Borazine itself is quite interesting and has earned some research of my own choice previously. For the preparation of the trinitrocompound I have two possible routes lined up, one route involves the reaction between silver nitrate and chlorotrimethylsilane (aka trimethylsilylchloride and trimethylchlorosilane) to produce the trimethylsilylnitrate which is a potent nitrating agent. Then the reaction of this in CH3CN with B-trichloroborazine and heating to drive off the volatile chlorotrimethlysilane. This reaction is a play on the reaction that was used to produce the B-triazidoborazine, however the stumbling block for this reaction is the aquisition of the chlorotrimethylsilane. Therefore my first attempts will simply be with B-Trichloroborazine in CH3CN then adding AgNO3 and hoping for the AgCl precipitate to confirm reaction. One other method that I found would be reacting borazine with anhydrous HNO3 @ 0C to form the -(BNO3HNH2)- compound then pyrolyzing that to form the desired product, however pyrolyzing something that in all likelihood will be a high explosive is a bad move in my book so I think I will try for the safer wet method.
Update 1/02/05
I recently found papers relating to the research of trinitro and trinitroso borazine by the use of silver nitrate and nitrite. Attempts similar to the ones that I devised, and they failed. However nitrotrimethylsilane was not used and might well provide interesting results, it was interesting to read, I will try to put excerpts from it here once I get some time.
Electrochemical Lithium Production from LiCl/LiBr
Just tinkering with this idea for now. As with all of my projects I have no use for the things I create other then to down the road create other things I have no use for. Still though I have found enough information in my sodium quest to make Li seem like an easy target, thus, I might take a shot at it. I might try a non-aqueous solvent or I might go for the LiCl/LiBr molten salt eutectic mixture.
Electrochemical Lithium Production from LiCl/KCl
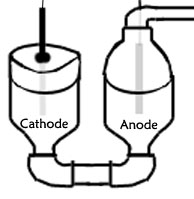
Recently I was reading up on methods of lithium production when I came across a practical example of this eutectic being used. And the good news is that it melts around 400 °C so fairly reasonable. Pictured above is my crappy drawing that I did of my cell to be. I consists of what is basically a metal U-tube made from pipes, a straight lenght of pipe, two 90° bends and two reducing/enlarging adaptors on top. This setup was tried some time ago in an attempt to electrolyze potassium hydroxide. It separates the products formed fairly well. Anyway, the components are liquified with a flame to be put beneath the center pipe. On the cathode side will be a metal endcap for a pipe, a hole will be drilled in the top an a steel cathode will be lowered in, it will be held in place by fireplace cement. On the other side will be another reducing/enlarging adaptor, so that the two large portions are touching one another. The smaller exit will be connected to some more pipes, preferably copper, to pipe the chlorine generated away, to a trap followed by an absorption train of NaOH. In the top of this system of pipes though a hole will be drilled so that a wire can be lead through, this will hang a graphite anode in the cell so that electrolysis can commence Because of the reactivity of the lithium produced the cathode side will be sealed throughout the experiment until everything has finally cooled down. It is my hope that things go right this time.
Tried non-aqueous mediums. Acetonitrile, acetone, nitrobenzene, mineral oil, kerosene, toluene, benzene, all gave no discernable results. DMSO however did. Electrolysis of a saturated LiCl solution in DMSO with nickel electrodes gave a flaky deposit on the cathode that reacted with water to form hydrogen. But the lithium reacts with the DMSO so yields are considerably low. Also the reaction smells.
Made some HBr (aq) so now I can neutralize some of my lithium carbonate to make LiBr and be one step closer to my eutectic mixture I need to create for molten electrolysis.
Side project for non-aqueous mediums (New 11/20/04)
There are a few lithium batteries out there that use organic carbonates as the electrolyte liquid in the cell. As such they have become one of my target molecules in the quest for lithium metal. Initially I thought that in order to make a chemical such as dimethyl carbonate I would need to react methanol with phosgene, now, phosgene is no where near a pleasant chemical to work with, so I was a little disdainful of this method, however thanks to one person at my board I now have a possible alternate procedure involving reacting urea with ethylene glycol in the presence of zinc oxide to give ethylene carbonate, the ethylene carbonate being separated from unreacted glycol by distillation at high temperature. If this is successful I will have a solvent that is known to 1) Conduct electricity with lithium ions and 2) Be stable to being in contact with lithium metal. So that is where this little project stands right now, going to get some high-grade anti-freeze and give this a shot.
This one I have a reason to want.... for the glory! I had to bone up on my knowledge of air conditioners and cooling in general but I have picked my method to try to liquefy air. The most accessible and easy to produce good results with method seems to be cascade cooling. You use the cooling effect of one liquid/gas to pre-cool another liquid in the next compressor system and allow it to liquefy with the pressure thus applied and when it vaporizes it absorbs even more heat and that liquid/gas in turn cools the next liquid/gas to an even lower temperature. My starting materials... eight air conditioners for houses. Been garbage picking lately and only have one so far but going for more. Too bad I have to replace the coolants in six of them and put an efficient desiccant over the air intake on the last one. Just everyone, cross your fingers and pray my electric bill doesn't sky rocket.
UPDATE : 6 - 25 - 04
I now have 3 air conditioners. All of them have had their motors gutted and one of them has an easy spigot on it to add or remove coolant. I have scaled down my plan and now only need 4 air conditioners total. The first air conditioner will use the regulation coolant, the second methyl chloride which I might have to make, the third will be CH2CH2 which I have no clue how to get. I could make it but I couldn't pressurize it or condense it. I need either a cylinder of it or I need to take the air conditioner to some specialist place and ask them to replace the coolant. Screwed either way. Those are the two coolants normally used in this cascade cooling procedure. The last air conditioner will need to be heavily modified however I am holding off on modifying any of them to see which air conditioner will be best suited for each part of the process.
Update (01/02/05)
Due to space constraints and the keeping of multiple air conditions, coupled with the frailty of the local power grid my attempts to liquefy air through cascade cooling are on hold indefinatley.
Hydrazine Production
-From catalytic decomposition of urea with a nickel catalyst
I have actually attempted this one but with dismal results. No encouragement whatsoever. The premise is that nickel is heated with urea and acts as a catalyst pulling out the CO and leaving just to haves of hydrazine, how convenient. Well, there may be two patents on it, but it doesn't seem to work too well. My run was with nickel turnings and pellets and my reaction was run all the way to 160C whereas the patent said it ran well at 40 - 120C. Even at 160C the reaction was not evident, and although bubbles were formed urea decomposes at these temperatures anyways. Still though I press on, another attempt will be made with nickel powder I just have to reduce some from solution. On my next try I may just try to collect a distillate, in my initial run I attempted to run the exit gasses though a chilled H2SO4 solution in an attempt to crystallize out any hydrazine sulfate formed to give a positive indication the reaction was working, no such luck.
-From partial oxidation of urea with Ca(OCl)2
This is just an attempt to modify the procedure for NaOCl to fit Ca(OCl)2 which is a more available/concentrated source of hypochlorite available in some areas. I tried some very preliminary tests and found some problems already, if one attempts to basify the Ca(OCl)2 solution you get Ca(OH)2 precipitate instead, however this leaves behind a NaOCl solution but that is not the intention of my attempts. I will attempt oxidation without basification and attempt to distill the azeotrope of hydrazine and water which is very near the hydrate in terms of mole composition. Originally I figured this would be a better method for the main reason that at the end of the reaction one could add a stoichiometrically calculated amount of H2SO4 and precipitate out all most all of the Ca2+ cation and therefore could sufficiently cool the resulting solution to obtain a maximum yield of hydrazine sulfate without worrying about the sodium sulfate crystallizing out all at once. But now I just want to get it to work.
My phosphorus attempts are on temporary hold so I won't be using my new reaction vessels or trying any of my other innovative mixtures I have planned. When I get back to work on it I want to try the "Make iron phosphide and heat that with sulfur" production method, sounds simpleish.
Ferrates
Electrochemical Preparation of:
Anodic oxidation of iron in highly concentrated alkali solution can lead to the formation of ferrates. I have taken several attempts at this from basically two different trains of thought.
Put a minimum amount of water into an iron crucible and put a larger volume of KOH in the same crucible as water. I put the electrodes beyond the moist KOH and into the highly saturated solution at the bottom. As electrolysis continued the solution heated up and more KOH went into solution. The all the KOH eventually solvated and the very hot solution turned black. After more electrolysis I dumped the solution into ice cold water with ice cubes floating in it. The solution was brick red and a precipitate settled to the bottom, later found to be iron from flaky deposits that started on the cathode under these conditions. Next I added a concentrated BaCl2 solution to the mix to precipitate the slightly soluble BaFeO4 I actually got precipitate, in one case a yellow-orange precipitate came out with some brick red mixed in which may have been Fe2O3. I also noticed that these solutions in ice cold water if not precipitated immediately would decompose very fast, 3 minutes or less. There was an evident color change and gas evolution. The solid precipitate that I ended up with reacted with HCl to produce a gas but the quantity was so low I was unable to ascertain the identity of the gas.
Other attempt was made with a beaker full of saturated KOH solution with a small amount of undissolved KOH remaining at the bottom. The anode was an iron rod an the cathode a nickel rod. Electrolysis immediately produced a red layer on the anode which did not produce near as much gas during the electrolysis, only an occasional bubble whereas the cathode looked to be boiling intensely. But that's about as far as the reaction went after several minutes. So I stepped it up a bit and replaced my iron anode with a large hunk of steel wool. Within 20 seconds there was an evident color change, a purple color that I had not saw when I tried electrolysis in the highly soupy mixture. The color change maxed out after 3 minutes or so so I discontinued electrolysis and prepared an ice solution to dump the contents into. However before I had the solution made up the color of the beaker had turned to an off yellow. Curious, I turned on the power again and let the solution change color. Then I turned it back off and started counting "One Mississippi, two Mississippi...." by the time 20 seconds had went by the color was once again a yellow derivative. Very fast decomposition even though the environment was very basic, although by now the temp of the mixture had risen from current. So I began electrolysis yet again and this time had my solution of BaCl2 made up. Dumping the now purple solution into the BaCl2 solution yielded initial cloudiness but a precipitate did settle out from it. The identical color of the precipitate I got from my previous experiments. While this was settling I started another run, I used the same technique but poured the mix into ice water without BaCl2, the solution remained purple and on addition of BaCl2 the solution still remained purple which was different then previous experiments, and I didn't get a precipitate, filtration took out the purple color but not as a solid, the color stuck in the cotton like a dye leaving a milky solution in the filtration flask. No tests were run on any of the products. But the solutions were always red before, never purple, possible manganese contamination? Or maybe I actually made the ferrate anion this time and the other times were invalid.
I attempted to make ferrate from further oxidation of iron (III) oxide. To do this I combined a large excess of oxide with NaNO3 and heated in an iron crucible. Classically a mix of about 50% NaNO3 and 50% NaOH are mixed for the oxidizing melt but I was wary of how the iron crucible would hold up to molten NaOH so I forwent that. After getting molten NaNO3 on my glove which caused it to immediately burst into flames, I dumped the melt on a large metal plate to allow it to cool. What I was left with was a brick red solid that broke easily, basically just iron oxide mixed intimately with NaNO3, tests proved that this experiment was a failure. My next run with be with NaOH in the mix in a good nickel crucible. Also, pyrotechnic mixtures of iron powder with excess NaNO3 yield ferrate with burned, purification would be the normal dissolve in cold water, precipitate with barium salt, filter.
Update 11/20/04:
I am either going to burn some of my sodium metal in an oxygen atmosphere to make sodium peroxide, buy it online for a high price, or make some barium peroxide or possibly buy it, then attempt to fuse it with ferric oxide to make a high yield of ferrate and attempt to study its properties better.
I have tried the charcoal/sulfur distillation method with very limited success and have been trying to come to a better way for some time. For a period I was thinking about a full setup utilizing the reaction between propane and sulfur vapor at 900C, which yields carbon disulfide and large amounts of H2S. The H2S was a problem but the engineering aspect was particularly a pain. Although not totally disbanded I have moved away from this idea and to a new train of thought.
In the laboratory H2S can be conveniently prepared by mixing sulfur with paraffin and glass wool and putting the mixture into a test tube, heating with a flame yields H2S. The process is the sulfur oxidizing the paraffin, H2S, the byproduct. Now, if that oxidization were allowed to go further it may well yield some CS2. But how to make it go further? More heat, but paraffins begin to volatize at temperatures between 350 and 450 C, to get them to go higher I need to go into longer carbon chains that will be less likely to volatize. Asphalt satisfies that, a very high boiling point, and long carbon chains, that should stick around long enough to give some CS2 upon heating with sulfur. So, I need to get some asphaltum, going to check my local hardware store and artist supply store, if no luck then I will try and online supplier, the stuff is dirt cheap.
Cyclic Ketone Peroxides in place of Crown Ethers
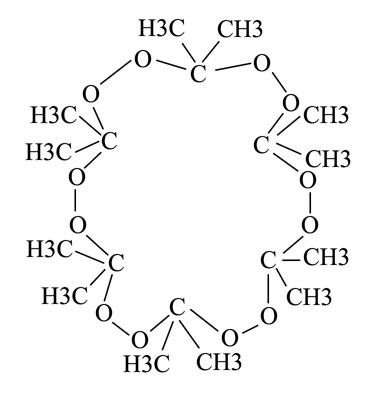
Just a kind of passing thought. Cyclic peroxides such as that commonly made from acetone not only look similar to crown ethers, but also possess an electron rich center which may be used to complex certain metals and pull them into non-aqueous solutions. Their physical characteristics are also similar, insoluble in water, soluble in some non-polar solvents, low melting point. They may on their own possess these solvating characteristics or it may be necessary to form them in the presence of the metal ions (Which is the way some of the crowns are made. They wrap around the metal ions to form the proper size and the complex is later decomposed leaving behind the crown.) If this does work then it would provide an extremely cheap alternative to crown ethers and open a whole realm for the home chemist. There may also be energetic aspects, such as complexing with a cation whose charge is balanced with an oxygen rich anion such as permanganate or perchlorate or persulfate. However my research will lean more toward the ability to pull ions into non-aqueous solutions.
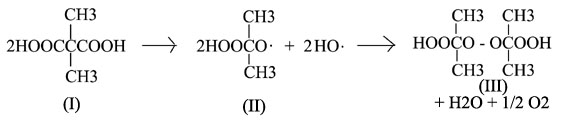
The normal tri compound being formed by the reaction of (III) with (I) present. (I got this specific reaction mechanism from the ACS journals). However with metal ions in solution, and slow drop by drop addition of H2O2 to acetone in neutral environments I may get something worthwhile. However the molecule itself is unhappy, as pictured further above, you can see the bond angles of the peroxide, normally it prefers linear I believe, however this case could cause it to derivate from that. In addition those methyl groups hanging out in the middle of nowhere are not favored, some would probably end up in the ring and may completely ruin any complexing effect if this compound could truly exist. Never the less these experiments seem like they would be easy to complete and I may give them a shot.
Preparation of THF using OTC Pentoses as the starting material.
Furfural and close derivatives can be prepared from pentoses by their reaction with acids. The online Organic Synthesis archives list the reaction between corn cobs and sulfuric acid with salt as their method of preparation of furfural. However the reaction between other pentoses should yield similar products, such as the reaction between OTC Xylitol and sulfuric acid with distillation. This should give a furfural derivative which can then be oxidized using a multistep treatment of the compound first with sodium hydroxide and then sulfuric acid. The resulting compound has to be decarboxylated which can occur at reasonable temperatures between 200 and 215 C, the furan produced distilling off, (I may just heat with DMSO and distill off the furan from that). Furan is an interesting chemical but it doesn't have a great shelf life and is prone to polymerisation, what I need to do is hydrogenate it to THF if that is at all possible. But what to use.... ?
Preparation of magnesium diboride (And subsequently diborane)
The reaction is simple, a Goldschmit type reaction between boric oxide an excess magnesium to give magnesium diboride. Going to perform this in a vessel with a one way exit gas valve, the resulting mixture of magnesium diborane, magnesium oxide, and unreacted magnesium and boric oxide is going to be difficult to separate without the help of water or acid so it's purification will depend upon the use intended. For example, diborane can reduce carboxylic acids to hydroxides, but borohydrides will not, so I could reduce say, succinic acid to the glycol and react that under basic conditions to give THF. But.... sounds like a pain in the butt. Another better method might be to react the diborane with a strongly basic solution to give borohydride which could be reacted with the furan produced above to give the saturated THF. The borohydride could also be made by reacting MgB2 directly with aqueous KOH however the product precipitates out and there is all that other insoluble stuff in there so... maybe that's not a good idea.
5-24-05
Earlier this month I tried this reaction, and very little borane was produced. After much researching I found that this reaction does NOT produce diborane and in fact produces a borane of higher molecular weight and less activity that must be distilled at liquid air temperature to give diborane. Also, according to the original work by Stock, huge amounts (>3 kg) of magnesium boride are used to give small amounts (a few liters of gas) of boranes, (incidentally it was found that the use of phosphoric acid increases yields of boranes produced from magnesium diboride). As such this reaction is not a good source for this reagent, therefore I am relying on a patent by which sodium or potassium borohydride is prepared by refluxing magnesium boride with NaOH or KOH in water. If potassium borohydride is obtained, its reaction with bromine or iodine in a polar aprotic solvent (many ethers or glymes) gives diborane if I am still interested in producing it.
From Calcium Cyanimide and HCl
Supposedly calcium cyanimide is available over the counter, however I have had no luck in picking it up in my area. So, time to make some. For this I have two possibilities, one is to react calcium hydroxide with urea at temperatures around 700C for some time, this can result in the formation of calcium cyanimide. Another method, the one I want to attempt, is to crush some calcium carbide up more and then put it in a vessel and force dry air through it. Calcium carbide will react with nitrogen straight from the air to yield calcium cyanimide. Once calcium cyanimide is made the plan is to react this with dry HCl gas at elevated temperatures resulting in the formation of ammonium chloride and carbon tetrachloride, hopefully at a temperature high enough to prevent much ammonium chloride form volatizing yet high enough that the CCl4 formed would be gaseous and make it to a conveniently placed condenser further down the line.
5-24-05
As has been brought to my attention (by Esplosivo from the sciencemadness discussion board), the reaction between calcium carbide and nitrogen only occurs appreciably at temperatures around 1000°C therefore that particular reaction has been removed as a feasible reaction.
7-29-05

Above is my home aid UV radiation apparatus produced by putting 70 UV LED's into a wooden bowl and running them in parallel. This was used in an attempt to chlorinate tetrachloroethylene to carbon tetrachloride. There were no useable results from the reaction because the crude material was disposed of.